
各位老师好,本期为大家带来单细胞转录组标准分析之降维聚类。
单细胞研究的重点就是对细胞进行分群和鉴定,然而单细胞测序数据是一个高维的复杂数据阵列,通常涉及到庞大的细胞数量,以及每个细胞中的众多基因。因此,面对复杂的数据阵列,在聚类之前,一般采用 PCA 方法进行适度降维以降低计算量和噪音,然后用 Leiden 方法寻找降维空间中邻近细胞网络的模块。最后,采用 tSNE 和 UMAP 两种非线性降维方法分别对单细胞群聚类结果作可视化分析展示。
Q:如何进行数据降维?
降维的过程其实就是去繁存简,每个基因对细胞来讲都是一个变化维度。数据特征维度太高,不但计算很麻烦,其次特征之间可能存在相关的情况 ,从而增加了问题的复杂程度,分析起来也不方便,所以需要尽可能保证真实差异的前提下减少维度的数量,PCA就是合理的方式之一,既可以减少需要分析的特征,也尽可能多的保留原来的数据信息。
步骤一:寻找高变基因
降维的过程依赖于基因表达量,因此挑选那些更能代表整体差异的基因进行降维分析是非常关键的,一般来说如果一个基因在细胞群体中变化幅度很大,它就是受关注对象,我们会认为是生物因素导致了这么大的差异,该基因即为高度变化基因(highly variable genes ,HVGs)。
Seurat包FindVariableFeatures函数,会计算一个mean-variance结果,根据基因表达量方差和均值筛选,以此获得高度可变的基因,一般默认采用前2000个高变基因进行后续降维分析。
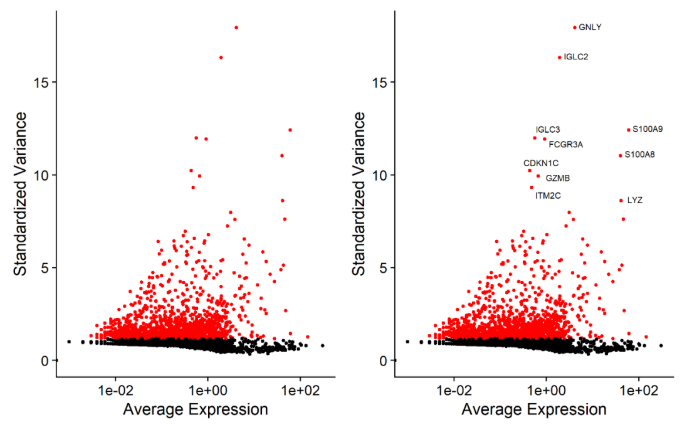
高变基因示例图
步骤二:降维
主成分分析 (PCA, principal component analysis)PCA的本质是将n个特征减少到k个,保留那些对生物差异贡献很大的特征。通过数据压缩,减少后面分析会使用到的维度,减少分析难度的同时尽可能的保留原有数据的特征。
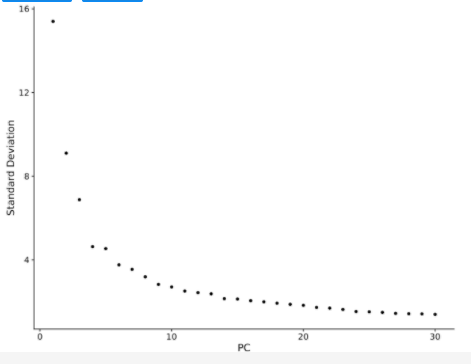
步骤三:选择合适的PCs
利用Elbow Point进行选择。Elbow Point作图后,一般选择斜率平滑的点之前的所有PC轴,加起来差异累计过90%就可以接受,根据后面聚类的结果可以重复调整。每个PCs都能捕获一些生物差异,而且前面的PC比后面的PC包含的差异信息更多,更有价值 。
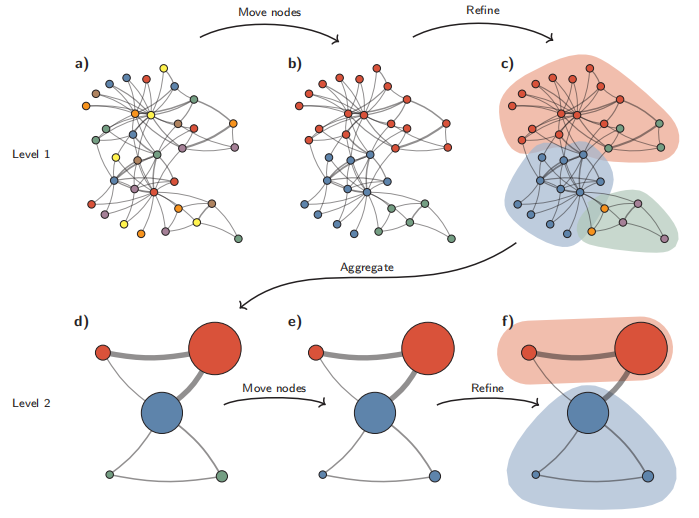
Leiden聚类算法原理图
Leiden算法从单例分区(a)开始。该算法将单个节点从一个社区移动到另一个社区,以找到合适的分区(b),然后对其进行细化(c)。基于细化分区创建聚合网络(d),使用非细化分区为聚合网络创建初始分区。例如,(b)中的红色社区被细化为(c)中的两个子社区,在聚合之后,它们成为(d)中两个独立的节点,都属于同一社区。然后,算法移动聚合网络(e)中的各个节点。在这种情况下,细化不会改变分区(f)。重复这些步骤,直到无法进行进一步改进。
聚类之后的可视化,目前主要有tSNE和UMAP。两者都是在高维空间中寻找保持相邻关系的低维表示方法。
Q:tSNE和UMAP的区别?怎么选择?
一是计算高维距离时,tSNE会计算所有点之间的距离,通过Perplexity(困惑度)参数调整全局结构与局部结构间的软边界,而UMAP则只计算各点与最近k个点之间的距离,严格限制局部的范围;另一方面,两种算法在对信息损失的计算方法不同,tSNE使用KL散度衡量信息损失,在全局结构上存在失真的可能,而UMAP使用二元交叉熵,全局和局部结构均有保留。目前两种方法在文献中均有使用,可根据实际情况来进行选择。