
研究背景
近年来,单细胞RNA测序(scRNA-seq)提高了我们对皮肤生物学的理解。但对新鲜样本的需求、不完全解离导致的细胞损失或死亡,以及组织消化过程中的刺激作用都使scRNA-seq的实用性受到了限制。而单核RNA测序(snRNA-seq)可以应用于冷冻的、难以解离的样本,这或许是可以绕过scRNA-seq对皮肤组织限制的一个途径。
研究目的
分析比对snRNA-seq和scRNA-seq在检测皮肤细胞时的效果差异。
研究方法
本研究对一个人的等分皮肤样本进行了snRNA-seq和scRNA-seq并行检测(单细胞测序技术服务由伯豪生物提供),并整合了先前发表的scRNA-seq数据,分析比较了两种方法在细胞比例和基因表达方面的差异。另外,通过Slingshot方法分析了角质形成细胞和成纤维细胞的分化轨迹。
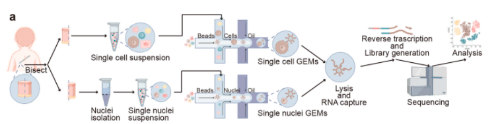
研究结果
1. scRNA-seq检测到数量更多的细胞和基因,尤其是免疫相关基因,而snRNA-seq受线粒体污染的影响较小,检测到更多种类转录因子。
scRNA-seq中定位到外显子区域的读数的比例为56.4%,显著高于snRNA-seq,其中读数更多地定位到内含子区域;scRNA-seq中的counts或基因数量高于snRNA-seq,但是snRNA-seq中每个细胞的线粒体读数的平均百分比低于scRNA-seq;对scRNA-seq和snRNA-seq中前100个高度可变基因的GO富集分析表明,snRNA-seq鉴定出更多与表皮发育相关的基因,而scRNA-seq则鉴定出更多的与免疫功能相关的基因;通过分析scRNA-seq和snRNA-seq检测到的转录因子的差异,发现snRNA-seq可以检测到更多的转录因子差异表达。
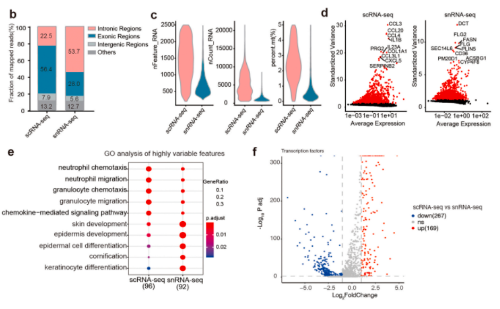
2 . snRNA-seq和scRNA-seq在检测细胞类型方面存在差异
由于scRNA-seq的不同消化方案可能导致细胞比例的变化,研究整合了已发表文章中健康皮肤的scRNA-seq数据进行比较分析。使用这些综合数据,鉴定出了13个细胞类型簇。研究使用已知的特征基因确定了每个簇的同一性;snRNA-seq对角质形成细胞和成纤维细胞的检测更有效,而scRNA-seq检测出更多种类的免疫细胞;消化时间和不同类型的消化酶对scRNA-seq中的细胞活性和基因表达有不同的影响。在He等人的数据中,用混合酶消化整个皮肤,与本研究的scRNA-seq和Reynolds等人的数据相比,获得了更多的角质形成细胞和角质形成细胞亚群。本研究和Reynolds等人在37°C下使用胶原酶IV过夜对真皮进行了消化,则获得了大量的免疫细胞和FIB1。相反,He等人的消化时间很短,成纤维细胞亚群和免疫细胞的百分比更接近snRNA-seq.
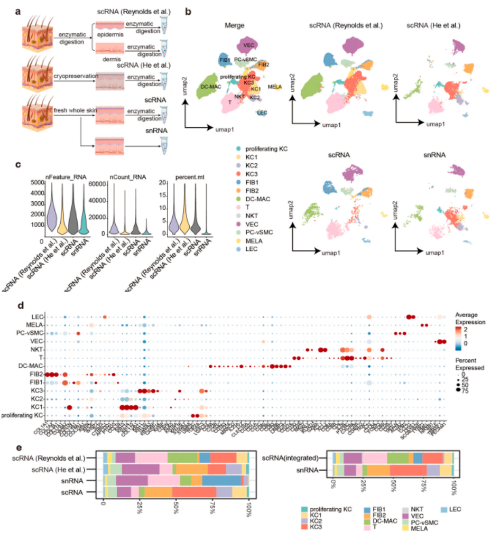
3. snRNA-seq揭示了更多与角质形成细胞功能相关的细胞簇
本研究基于snRNA-seq的结果,对角质形成细胞进行了二级无监督聚类,并通过特征基因和GO富集分析鉴定了8个亚聚类。皮肤形态发生相关的“基底”(高表达COL17A1、KRT15和KRT14)细胞;角质形成细胞分化和角质形成相关的“棘状”(高表达KRT1和中等表达KRT10)和“颗粒状”(高度表达LOR、FLG和SPINK5);参与细胞连接组装的 “通道间隙”(表达GJB2和GJB6)细胞;ATP代谢,调节细胞连接的离子通道相关的“通道ATP酶”(表达ATP1A1和P1B1)细胞;角质形成细胞分化和细胞连接的过程相关“毛囊(外根鞘)”(表达KRT6A、KRT6B和KRT17)细胞;脂肪酸代谢以分泌皮脂相关的“毛囊(内鞘(IR)-皮脂腺)”(表达KRT79、ELOVL5和FASN)细胞;有丝分裂核分裂相关的“有丝分裂”(表达UBE2C、TOP2A和MKI67)。这些亚群很好地概括了角质形成细胞的空间和功能聚类;利用snRNA-seq中角质形成细胞亚群的分群方法对scRNA-seq数据进行处理,发现两种方法中,角质形成细胞子群的比例不平衡。在scRNA-seq数据集中,功能性角质形成细胞的比例显著降低,包括“通道ATP酶”、“通道间隙”、“棘状”和“有丝分裂”细胞。
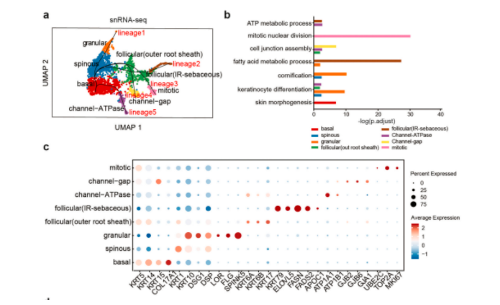
4. snRNA-seq鉴定出调控角质形成细胞分化的基因
由于角质形成细胞的分化状态与亚群密切相关,本研究通过伪时间分析探究了角质形成细胞分化过程。“基底”细胞被定义为起点,五个谱系被识别为代表不同的轨迹。沿着这些轨迹,本研究发现了代表角质形成细胞分化特定阶段的新标志物。例如,LYPD3和EMP2的表达随着角质形成细胞的角质化而增加;CSTB主要在毛囊外根鞘周围的角质形成细胞中表达;结合人类蛋白质图谱(HPA)数据库,证实了这些基因在原位皮肤中的蛋白质表达与转录组分析一致;“基底”和“毛囊(外根鞘)”细胞高表达NFIB,用于维持干细胞特性并调节细胞周期;GRHL1在分化轨迹末端表达逐渐增多,表明其参与角质形成细胞的功能分化。在HPA数据库中,NFIB主要在正常皮肤和基底细胞癌(BCC)的“基底”角质形成细胞中表达。GRHL1在正常皮肤的基底上角质形成细胞中表达,但在cSCC中不表达。

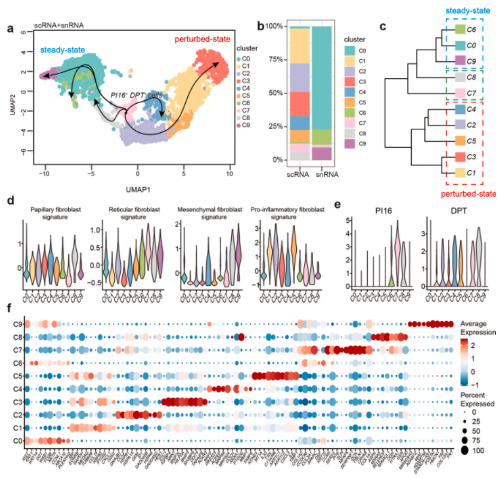
5. snRNA-seq揭示稳态下的人类成纤维细胞图谱
由于成纤维细胞在解离过程中会发生显著变化,本研究在集成数据集上进行了二级无监督聚类,进一步的确定了其异质性;与scRNA-seq相比,snRNA-seq中的亚簇数量显著较低;根据位置和功能(分泌性乳头状细胞、分泌性网状细胞、间充质细胞和促炎细胞),并结合最近提出的四个亚群的特征基因,进一步定义了成纤维细胞的亚群;促炎性成纤维细胞的基因表达特征主要局限于C1-C5中的成纤维细胞,且仅在scRNA-seq数据中发现,表明酶消化可能激活成纤维细胞并诱导炎症表型。C1和C3具有乳头状成纤维细胞的特征;C2主要表现为网状成纤维细胞的特征;C4和C8主要表现出间充质特征。C0、C6和C9主要存在于snRNA-seq数据中。这三个亚群共享Pi16+成纤维细胞祖细胞。C7高表达PI16和DPT,这是泛成纤维细胞祖细胞的两个标志物,并被选为成纤维细胞轨迹分析的起点。Slingshot谱系推断确定了从PI16+簇中出现并在两个方向上结束的五个轨迹:稳定簇或激活簇,表示皮肤成纤维细胞在稳定或激活状态下的分化轨迹。状态特异性特化成纤维细胞与分级聚类分析完全一致,表明scRNA-seq中的单细胞分离可能导致成纤维细胞的炎症激活,而snRNA-seq反映了成纤维细胞在原位的稳定状态。
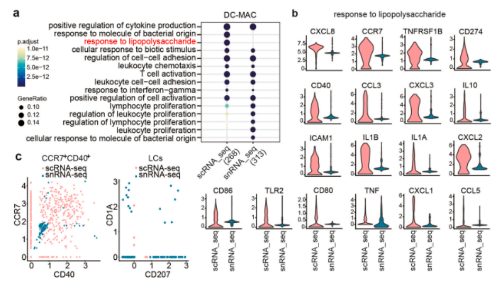
6. scRNA-seq和snRNA-seq在识别皮肤中的免疫细胞方面是互补的:scRNA-seq具有更大的检测免疫细胞的能力;而snRNA-seq在检测某些稀有细胞方面更为优越
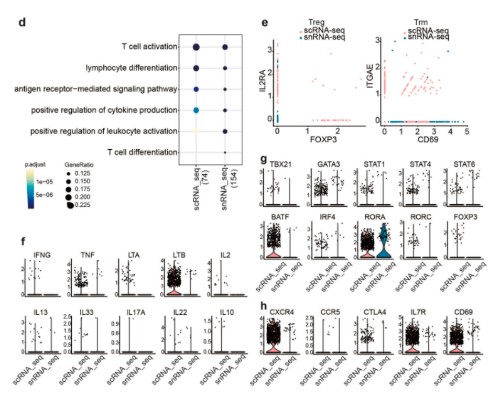
本研究分析了包括DC、巨噬细胞和T细胞在内的主要免疫细胞基因表达的变化。scRNA-seq和snRNA-seq都揭示了DC和巨噬细胞中常见的抗原呈递细胞(APC)功能,包括细胞因子产生、白细胞趋化性和T细胞活化的阳性调节。然而,scRNA-seq展现出更多的免疫相关功能,如“对脂多糖的反应”,并且该GO术语中的大多数基因编码有效或功能性分子。scRNA-seq还鉴定出比snRNA-seq数量高得多的CCR7+CD40+迁移APC,但几乎没有检测到任何LC;而snRNA-seq可以鉴定少量的LC;研究还发现,scRNA-seq和snRNA-seq都检测出大量与T细胞功能相关的生物学过程。其中,scRNA-seq鉴定了罕见的驻留T细胞。编码分泌细胞因子、表面标记物和转录因子(RORA除外)的T细胞亚群相关基因的丰度在scRNA-seq数据集中远高于snRNA-seq。
研究总结
在对皮肤组织的研究中,snRNA-seq不太容易受到线粒体和核糖体RNA的污染,并能检测到更多种类的转录因子;通过这些有别与scRNA-seq的结果,可以对细胞亚群的功能性进行更深入的研究。相较于scRNA-seq的组织解离带来的刺激,snRNA-seq可以检测出稳态环境下的转录组特征。在检测免疫细胞方面,scRNA-seq具有更大的检测免疫细胞的能力;而snRNA-seq在检测某些稀有细胞方面更为优越。
原文链接:
https://www.jdsjournal.com/article/S0923-1811(23)00145-7/fulltext